はじめに
前編ではFASTAファイルを読み込む部分のプログラムを作成しました。後編ではOpenBabelを利用してPDBファイルの残基番号の修正をします。プログラムに入力するデータなどは前編で紹介したものを利用する仕様になっています。また、今回のプログラムは前編で作成したプログラムをそのまま利用しています。したがって、前編を読んでからこの後編を読んで頂いた方が良いと思います。
プログラム解説
今回のプログラム(プログラム名をrenumbering.cppとします)は前編でclustaloコマンドで作成したFASTAファイル(aligned.fasta)とMOL2(1lke.mol2)ファイルを読み込んで、残基番号が修正されたPDBファイル(rev_1lke.pdb)を出力します。ここでMOL2ファイルは以下のOpenBabelのコマンドで作成します。
$ obabel 1lke.pdb -omol2 -O 1lke.mol2MOL2ファイルに変換する理由は、PDBファイル中の原子座標に関する情報以外を一旦リセットするためです。例えばアミノ酸配列中の何処から何処までヘリックス構造であるという記述もあるのですが、OpenBabelではこちらの方までは修正してくれません。この情報が残っているとPDBのビューアーで表示させたときに、変な構造となる場合があるのです。仕方ないのでMOL2形式に変換することで消去しています。
このプログラムのコンパイル方法は以下の通りです。
$ g++ -std=c++11 Protein-Ligand.cpp \
-I${HOME}/anaconda3/envs/ob3/include/openbabel3 \
-L${HOME}/anaconda3/envs/ob3/lib \
-Wl,-rpath=${HOME}/anaconda3/envs/ob3/lib -l openbabela.outというファイルが出来ていれば成功です。プログラムの実行は以下のコマンドです。実行するとrev_1lke.pdbというファイルが作成されているはずです。
$ ./a.outそれではプログラムを見ていきましょう。C++のコードは以下の通りです。
1 #include <iostream>
2 #include <fstream>
3 #include <sstream>
4 #include <string>
5 #include <vector>
6 #include <map>
7
8 // OpenBabel
9 #include <openbabel/mol.h>
10 #include <openbabel/residue.h>
11 #include <openbabel/obconversion.h>
12
13 using namespace std;
14 using namespace OpenBabel;
15
16 // 1文字を3文字に変換するための変数設定
17 map<char, string> aa3;
18
19 void init(void) {
20 aa3['A'] = "ALA";
21 aa3['R'] = "ARG";
22 aa3['N'] = "ASN";
23 aa3['D'] = "ASP";
24 aa3['C'] = "CYS";
25 aa3['E'] = "GLU";
26 aa3['Q'] = "GLN";
27 aa3['G'] = "GLY";
28 aa3['H'] = "HIS";
29 aa3['I'] = "ILE";
30 aa3['L'] = "LEU";
31 aa3['K'] = "LYS";
32 aa3['M'] = "MET";
33 aa3['F'] = "PHE";
34 aa3['P'] = "PRO";
35 aa3['S'] = "SER";
36 aa3['T'] = "THR";
37 aa3['W'] = "TRP";
38 aa3['Y'] = "TYR";
39 aa3['V'] = "VAL";
40 aa3['-'] = "GAP";
41 aa3['X'] = "ANY";
42 }
43
44 class SEQ {
45 string name;
46 string seq;
47 vector<pair<string, int> > seq3l;
48 int pos;
49
50 public:
51 SEQ() : pos(0) {} // コンストラクタ
52
53 SEQ(const SEQ& Obj) { // コピーコンストラクタ
54 name = Obj.name;
55 seq = Obj.seq;
56 seq3l = Obj.seq3l;
57 pos = Obj.pos;
58 }
59
60 ~SEQ() {} // デストラクタ
61
62 bool ReadFASTA(istream* ist) {
63 bool _f = false;
64 string line;
65 pos = 0;
66
67 name.clear();
68 while (getline(*ist, line)) {
69 if (line[0] == '>') {
70 name = line.substr(1);
71 _f = true;
72 break;
73 }
74 }
75 if (!_f) {
76 return false;
77 }
78
79 seq.clear();
80 streampos spos;
81 while (getline(*ist, line)) {
82 if (line[0] == '>') {
83 break;
84 }
85 seq = seq + line;
86 spos = ist->tellg();
87 }
88 ist->seekg(spos);
89
90 this->ToTLC();
91
92 return (true);
93 }
94
95 void ToTLC(void) {
96 seq3l.clear();
97 for (int i = 0; i < seq.size(); i++) {
98 seq3l.push_back(pair<string, int>(aa3[seq[i]], i + 1));
99 }
100 }
101
102 string GetName(void) { return name; }
103
104 void SetName(string s) { name = s; }
105
106 int length(void) { return seq.size(); }
107
108 void Reset(void) { pos = 0; }
109
110 char GetChar(int p) {
111 if (p > seq.size()) return '\0';
112 return seq[p - 1];
113 }
114
115 string GetTLC(int p) {
116 if (p > seq.size()) return "END";
117 return seq3l[p - 1].first;
118 }
119
120 pair<string, int> Next(void) {
121 if (pos >= seq.size()) return pair<string, int>("END", pos);
122 return seq3l[pos++];
123 }
124
125 void print_seq(ostream* ost) {
126 string x;
127 *ost << ">" << name << endl;
128 int i = 0;
129 for (; i < seq.size() / 60; i++) {
130 x = seq.substr(i * 60, 60);
131 *ost << x << endl;
132 }
133 x = seq.substr(i * 60);
134 *ost << x << endl;
135 }
136 };
137
138 int main(int argc, char* argv[]) {
139 // 初期化
140 init();
141 SEQ s;
142
143 ifstream ifasta("aligned.fasta");
144 s.ReadFASTA(&ifasta);
145 ifasta.close();
146
147 ifstream ifs("1lke.mol2");
148 ofstream ofs("rev_1lke.pdb");
149
150 // フォーマット設定
151 OBConversion conv;
152 conv.SetInAndOutFormats("MOL2", "PDB");
153
154 // 分子の読み込み
155 OBMol mol;
156 if (!conv.Read(&mol, &ifs)) {
157 cout << "error!" << endl;
158 exit(1);
159 }
160
161 pair<string, int> tlc;
162
163 s.Reset();
164 for (OBResidueIterator rr = mol.BeginResidues(); rr != mol.EndResidues(); rr++) {
165 do {
166 tlc = s.Next();
167 } while (tlc.first == "GAP");
168 if (tlc.first == "END") break;
169
170 string r = (*rr)->GetName();
171 r=r.substr(0,3); // 最初の3文字を切り出す
172 // アミノ酸が一致した場合に番号変更
173 if (r == tlc.first) {
174 cout << (*rr)->GetName() << "\t" << (*rr)->GetNum();
175 cout << " -> " << tlc.second << endl;
176 (*rr)->SetNum(tlc.second); // 番号変更
177 }
178 }
179
180 conv.Write(&mol, &ofs);
181 ofs.close();
182
183 return 0;
184 }それでは簡単に説明しましょう。最初から136行目までは前編で紹介したプログラムとほとんど同じです。9〜11行目にOpenBabelに関するヘッダーファイルをincludeしている部分が異なるだけです。ここで、アミノ酸残基を扱うためにresidue.hというヘッダーファイルがあることに注意してください。特にOpenBabelの古いバージョンではmol.hの中にresidue.hも含まれていたので、このヘッダーファイルをincludeする必要は無かったのですが、新しいOpenBabelのバージョン3ではこのように明示的にincludeしないとコンパイルエラーになってしまいます。私はバージョン3のOpenBabelを初めて利用したときに、この仕様に気付かずにハマりました。オープンソースだから仕方ないのかもしれませんが、突然仕様変更するのはやめて欲しいものです。
138行目からmain関数が始まり、143〜159行目まではFASTAとPDBファイルを読み込んでいます。161行目のtlcという変数の宣言はFASTAファイルの各アミノ酸の要素を受け取るためのものです。pair型のfirst要素はアミノ酸の3文字表記が格納され、second要素には番号が格納されます。
164行目から177行目までのループでPDBファイル中の残基番号の修正をしています。これはPDBファイル中のアミノ酸をイテレータを利用して順番に処理をするループになっています。このループ中の165〜167行目ではFASTAファイル中のGAPを読み飛ばしています。168行目でのif文はFASTAファイルが終了したときにループを抜けるためのものです。170行目でPDBファイル中のアミノ酸の3文字表記を取得しています。そしてFASTAファイル中のアミノ酸と一致した場合に、176行目で残基番号を変更しています。最後に180行目で修正されたPDBを出力してこのプログラムは終了です。
計算結果
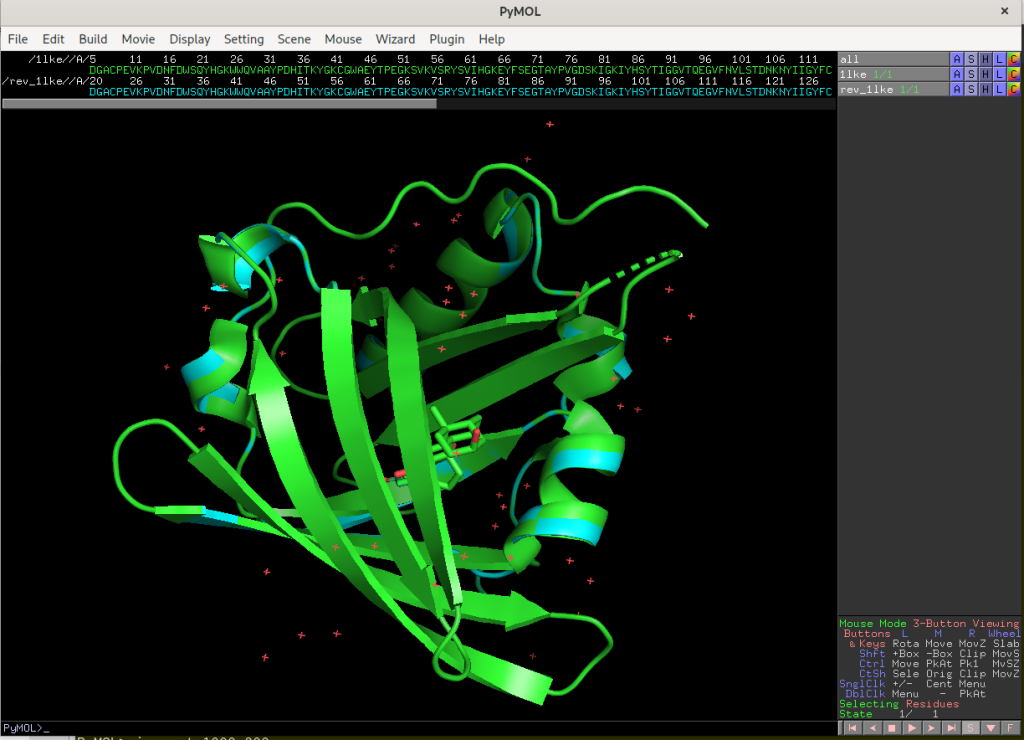
以下の図は1lke.pdbとrev_1lke.pdbをPyMOLで開いたときの画面です。上のアミノ酸番号(シアン色)を見ると番号が変更されていることがわかります。
Category: OpenBabel, プログラミング関連